- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
Introduction to the SARS-CoV-2 Genome Browser
The UCSC Genome Browser is an open-source, interactive sequence visualization tool that has been a cornerstone of genomics since we released the first human genome assembly 20 years ago. Cited in more than 37,000 scientific articles and used by thousands of researchers each day; it allows for cross-referencing of research, clinical, and epidemiology data against reference genomes, including SARS-CoV-2. This data is continuously updated and added to as new datasets become available. For a more thorough description, please reference our SARS-CoV-2 Genome Browser Nature Genetics paper. We also post updates and COVID Browser resources to out COVID-19 Browser home page.
This guide will go through some of the most important use cases of the SARS-CoV-2 Genome Browser. These topics include:
- Orientation and Navigation
- Gene Data and Sequence Alignments
- Variation and Immunology data
- Phylogenetic Contact Tracing using UShER
- Other tools and data downloads
- Support and Collaboration
For those who prefer a video explanation, we also have the following tutorial:
Genome Browser Orientation and Navigation
The standardized reference genome displayed on the COVID Genome Browser is from one of the first isolated cases, known as NC_045512v2 or wuhCor1. With more than 80 track datasets across the SARS-CoV-2 reference genome's nearly 30,000 RNA bases, navigation is essential to finding the information you want to see. Below is an example view of the SARS-CoV-2 Genome Browser with labeled sections highlighting the navigation, reference sequence, annotations, and other available track datasets.
Navigation controls at the top allow users to move left and right and to zoom. The search box allows users to search for particular features or to move to exact genomic coordinates. The RNA sequence is shown at the top only when the view is sufficiently zoomed in. Annotations are shown for data tracks that have been set to visible in the available tracks section at the bottom. Tracks can be configured with a right-click or by clicking on their name near the bottom of the page.
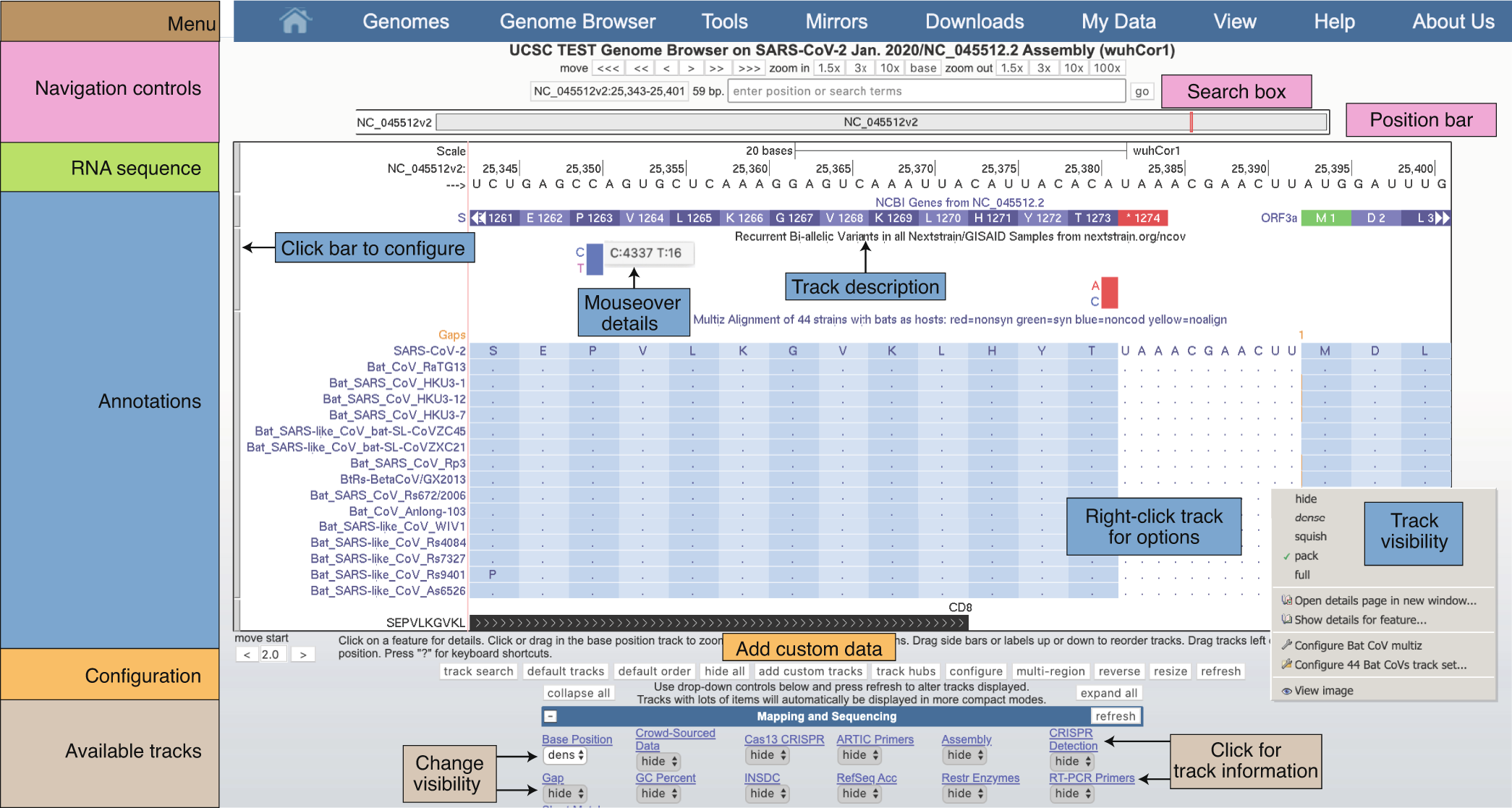
This is a view of the SARS-CoV-2 Genome Browser (COVID Browser) with labeled elements to help with orientation. Interact with this session by clicking on the picture. To read the full caption, please go to our Nature Genetics paper.
Genes and Sequence Alignments
Gene and protein annotations are organized by the contributor, most notably NCBI and UniProt. Having multiple information sources allows a consensus to be formed among datasets. Like many viral genomes, molecular complexity arises from polyproteins rearranging, generating ~29 protein products. Most notable among these is the S (spike) protein which defines coronaviruses and allows entry into cell membrane. Additional tracks contain information such as interactions between viral proteins and human proteins (protein interact), PDB structures, and RNA structure annotations (Rangan RNA), and more.
Sequence alignments and conservation data are also available across the SARS-CoV-2 genome, from large-scale views to individual bases and amino acids. Four conservation tracks compare sequences with 44 bat coronaviruses, 119 vertebrate coronaviruses, 7 human coronaviruses, and PhyloCSF computed conservation scores. The tracks display differently depending on visibility mode and the number of bases on the screen.
Datasets can be turned on by setting the dropdown next to the data track name from "hide" to dense, squish, pack, or full. Then click the button to see these changes in effect. Clicking on a data track name will take you to a description with more information on the dataset, display conventions, methods, and references. Clicking on a particular item will take you to a page with complete information about that item and dataset.
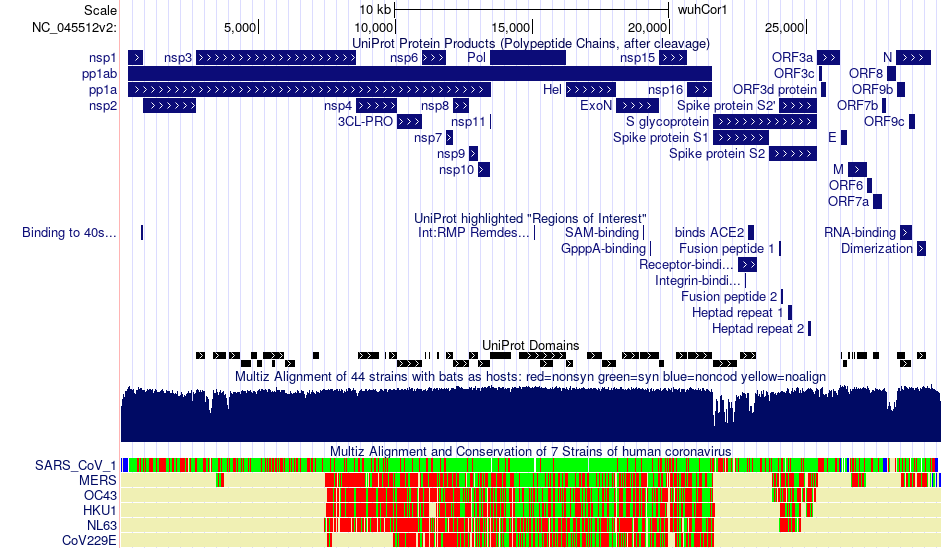
This Genome Browser display shows some of the gene and conservation tracks available on the SARS-CoV-2 genome. You should be able to see UniProt protein products, regions of interest, and domains all mapped against the SARS-CoV-2 genome. Below those tracks are two different conservation alignments in "squish" and "pack" formats, comparing bat-host and human-host coronavirus sequences with the reference SARS-CoV-2 genome. Interact with this session by clicking on the picture.
Exploring Variation and Immunology Data
The SARS-CoV-2 Genome Browser displays data on variation within SARS-CoV-2 from UniProt, GenBank, GISAID, Nexstrain, and other providers. These datasets cover global trends in SARS-CoV-2 variation among all available public sequences, with regional descriptions available through clicking into a particular entry. A few of the most notable tracks under the "Variation and Repeats" section are the Phylogeny: Public track, which shows a continuously updating phylogenetic tree that clusters similar sequences, with the frequency of each mutation shown by the height of the bar at that particular base. Tools are provided to filter these data to show only well-supported mutation calls, set thresholds for minor-allele frequency, and display data for specific clades.
Another track is the spike protein mutations from community annotations, highlighted as amino acid changes with red indicating strong antibody escape in receptor-binding domain (RBD) mutation screens. The Genome Browser has also has the Variants of Concern track, which pinpoints each accumulated mutation that defines 4 strains of SARS-CoV-2 of particular concern, labeled based on lay terms (such as 'California variant') as well as the using the lineage defined by the Pangolin software (such as 'B.1.1.7').
The Genome Browser also provides 12 immunology datasets that can inform potential therapeutic targets or public health risks. Protein epitopes are highlighted in the genome by multiple tracks, including those from the Immune Epitope Database (IEDB) and from a study of COVID+ patients. Of particular interest are the datasets describing surveys of antibody response across a variety of SARS-CoV-2 variants in the receptor-binding domain (Antibody Escape Mutations).
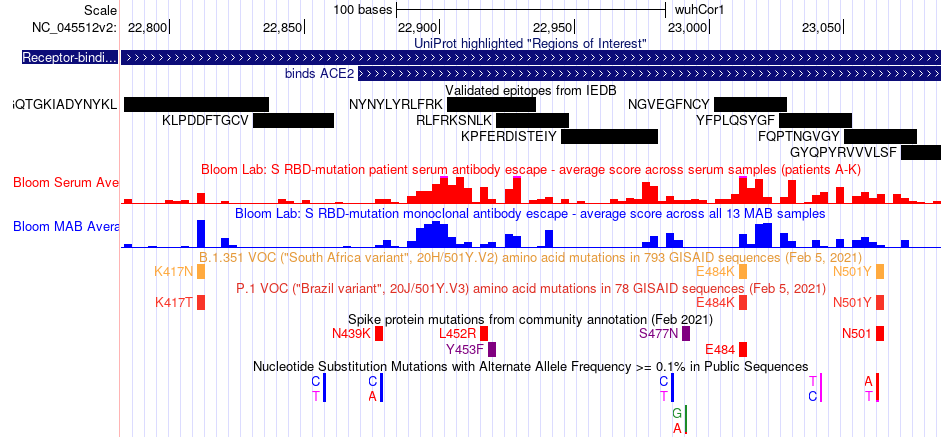
This image shows some of the variation data tracks that can be displayed on the SARS-CoV-2 genome, specifically zoomed into the receptor-binding domain of the Spike protein. Validated epitopes are displayed in black that may be a target for therapeutic antibodies. In red and black, antibody escape scores are are shown for each genome position. Smaller tick marks show amino acid or nucleotide changes from different sources, with more information available by clicking into the item.
Genetic Contact Tracing with UShER
The UCSC Genome Browser has developed a tool that allows placement of SARS-CoV-2 sequences onto existing phylogenetic trees far faster than previous methods, allowing instantaneous tracing of strains and transmission events. This tool is called Ultrafast Sample placement on Existing tRees (UShER) and exists as an interactive web-tool to compare sequences and link to existing public phylogenetic trees.
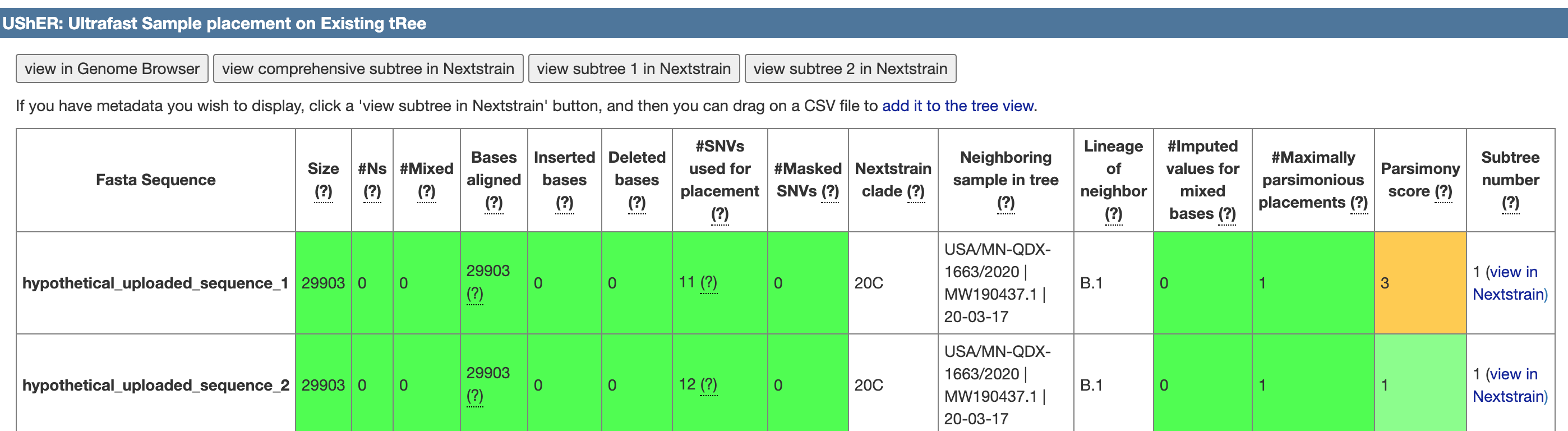
After uploading a Fasta file, the tool returns a page with quality metrics such as: number of bases aligned, number of Ns, and number of maximally parsimonious placements along with the lineage and clade of the nearest neighbor. Colored boxes highlight possible quality issues, green meaning this was a high confidence placement.
SARS-CoV-2/ COVID Phylogenic Trees
You can view your aligned SARS-CoV-2 sequence genotypes along with their closest known relatives among the 150,000+ public sequences. You can compare among your uploaded samples or trace possible transmission vectors using mutational signatures.
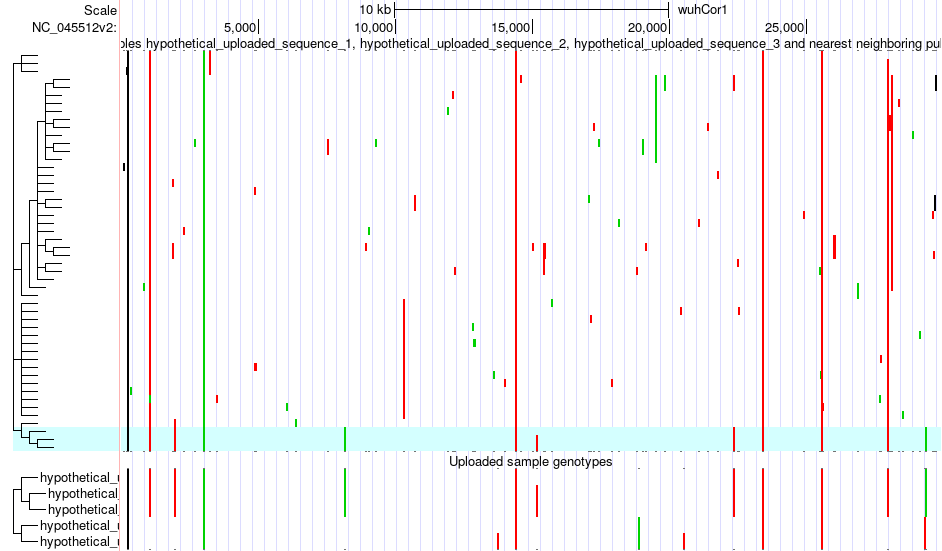
The uploaded sequences are highlighted in blue alongside their most closely aligned public sequences. You can investigate genotypes and relationships between samples.
Other tools, downloads, and features
Custom Tracks, BLAT, Track Hubs
Along with a suite of data tracks, filters, and visualization options for the SARS-CoV-2 genome, the UCSC Genome Browser offers many additional ways to interface with our data. You can upload your data on the reference genome in nearly any format with our Custom Track tool. If you have unaligned sequence, you can use our BLAT sequence alignment tool to get coordinates and base-by-base comparison with any reference genome. We also display formatted data as Track Hubs and curate a list of user-submitted Public Track Hubs.
Downloads, Table Browser, JSON API, SQL
As part of our open-source, open-access philosophy, we try to make it as easy as possible for researchers to download entire datasets or filtered subsets. Each track description page has a Data Access section which points users to our main options for data download. For downloading complete datasets, our SARS-CoV-2 download directory provides access to all our source files for transparency and reproducibility. Our Table Browser tool lets users interact with our data using a variety of filters based on score, identifiers, or any other field. Table Browser also allows users to convert data into multiple different formats (e.g. BED, GTF) and to access different formatted sequence outputs (in FASTA format).
We have a JSON API which can be programmatically called and return any dataset in its entirety or as a filtered subset based on documented input parameter. We also offer a Public SQL server for similar flexible, automatic way to access genomic data and annotations. Along with this particular virus genome browser, we have thousands of genomes available for visualization and analysis from our genome assemblies gateway page.
Support and Collaboration
The Genome Browser offers rapid email support for anything related to our tools. If your question is general or may have been asked before, please review our Browser documentation and our archive of previously answered questions. If you would still like help, please go to our Contact Us page to see access our email support. When contacting us, please include a session link, images, and example data if applicable. We are active on social media, you can follow us on Twitter or Facebook.
We are always looking to collaborate with researchers and add new datasets to our site. We also seek to continuously improve our tools to meet the needs of the scientific community. If you have any collaboration ideas, contributions, or feature requests, please reach out through our suggestion page.